Scleroderma (literally “hard skin”) is an umbrella term for a family of rare diseases with the common factor being abnormal thickening (fibrosis) of the skin. However, not everyone with scleroderma develops skin changes. With some variants of the disease, skin changes usually occur early in the disease process and can develop very rapidly. With other forms of scleroderma, skin changes may not occur for many years after the development of other symptoms and in rare cases may never be a significant symptom of the disease.
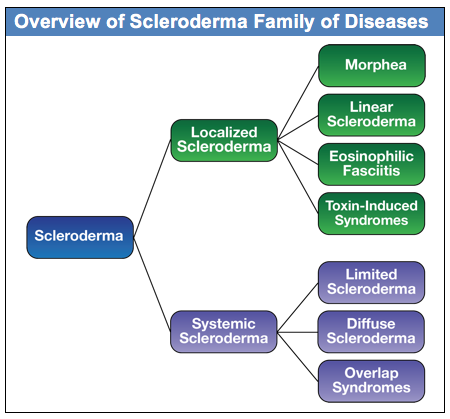
There are two main groupings of the scleroderma family of diseases: Localized and Systemic, as shown in this diagram. Generally speaking, the localized forms of scleroderma are limited to different kinds of skin changes and do not have internal organ involvement. In contrast, the systemic forms of scleroderma are complex autoimmune diseases that affect organs throughout the body in addition to skin changes. The focus of both the Scleroderma FAQ and this Guide is on the systemic forms of scleroderma, although basic information is included in the FAQ on the localized forms of scleroderma.
Within the systemic forms of scleroderma, there are three major categories of the disease: diffuse, limited and overlap syndromes. The more rapidly progressing forms of systemic scleroderma are in a category called diffuse scleroderma. In research literature, this is referred to as diffuse cutaneous systemic sclerosis and is commonly abbreviated as dcSSc. This form of systemic scleroderma is typically characterized by rapid development of skin thickening, beginning with the hands and face and extending to the arms and trunk. People with diffuse scleroderma are at greater risk for developing internal organ involvement early in the disease process. The specific internal organ systems that are affected depends to some degree on which specific type of diffuse scleroderma the patient has, as often indicated by the patient’s antibody profile.
The second major category of systemic scleroderma is called limited scleroderma. The word “limited” refers to the fact that the skin involvement in this form of systemic scleroderma is usually limited to the lower arms and legs and sometimes the face. There is still significant internal organ involvement with limited scleroderma, but it generally develops more slowly than with the diffuse form. In research literature, this is referred to as limited cutaneous systemic sclerosis and is commonly abbreviated as lcSSc. It is worth noting that this form of scleroderma used to be referred to as CREST Syndrome, and you will still find many articles that use the older term. While limited scleroderma progresses more slowly and has a better overall prognosis than diffuse scleroderma, different variants of limited scleroderma (based on antibody profile) have different complication risks over the long term.
The third category of systemic scleroderma is a diverse group that is generally referred to as scleroderma overlap syndromes. With overlap syndromes, while patients have clear scleroderma specific symptoms, they also have symptoms that overlap with other autoimmune diseases including lupus and myositis (muscle inflammation). An example is Mixed Connective Tissue Disorder, which includes symptoms that are common in scleroderma, lupus, and myositis. The specific antibody determines the nature of the overlap syndrome.
Affected Population
Recent studies estimate that in the US the incidence of new cases is about 20 per million adults (about 4800 new cases per year based on current US population estimates) and that the current prevalence is about 240 cases per million adults (about 60,000 total active cases). The American College of Rheumatology estimates that the number may be as high as 100,000 people in the US. A number of international studies suggest that scleroderma occurs much more frequently in the United States than elsewhere. These regional differences may be a consequence of differential genetic susceptibility to scleroderma, different exposure to possible environmental triggers, different diagnostic criteria, or a combination of these factors.
Scleroderma may occur at any age, but the symptoms most frequently begin in mid-life (25-45). The diffuse and limited forms of scleroderma are very rare in children. The disease is about 4 times more common in women than men.
There seems to be a relatively weak genetic link with scleroderma. Close order relatives of an affected individual are more likely to have some type of autoimmune condition but this is more likely to be a different disease, such as rheumatoid arthritis, Hashimoto’s (autoimmune hypothyroidism), Graves (autoimmune hyperthyroidism), or lupus. Also, close order relatives of affected patients may have elevated anti-nuclear antibody (ANA) levels as compared to the normal population, but without any symptoms of any autoimmune disease.
Causes
The exact cause of scleroderma is unknown. There are a number of environmental factors that appear to be related to scleroderma or scleroderma-like illnesses, including exposure to silica dust, vinyl chloride, epoxy resins, and other organic solvents. Several studies have shown some evidence of geographic clustering, which is also consistent with possible environmental risk factors. Scleroderma is best thought of as a disease with two components: genetic susceptibility and a trigger event, for example, exposure to silica dust or organic solvents.
There is some research support for the idea that a subset of scleroderma patients may have systemic infections as a possible trigger for their scleroderma. While there have not been any studies directly linking Lyme disease to scleroderma, there is some research that suggests that Lyme disease may be a possible trigger for scleroderma in susceptible patients.
Symptoms
One important thing to realize with scleroderma is that while there is some commonality of symptoms for different types of scleroderma, there is a tremendous variability in terms of which symptoms ultimately will occur and in what order.
Scleroderma often begins with Raynaud’s phenomenon – the fingers and sometimes the toes lose circulation and turn white upon exposure to cold. Raynaud’s phenomenon usually (but not always) precedes skin changes by several months with diffuse scleroderma and often precedes skin changes by several years with limited scleroderma. Other early symptoms may be painful joints, morning stiffness, red swollen hands, fatigue, and/or weight loss.
It is important to note, however, that Raynaud’s phenomenon without any underlying disease is not uncommon in the general population, especially among young women. This form of Raynaud’s is called “primary Raynaud’s.”. A key distinguishing characteristic is that with primary Raynaud’s, the anti-nuclear antibody (ANA) will normally be negative, while with Raynaud’s which accompanies scleroderma or other auto-immune disorders (secondary Raynaud’s), ANA is usually positive. The vast majority of young women with Raynaud’s symptoms that appear in their teenage years never develop a positive ANA or any systemic damage or skin changes. However, in a small percentage of this population, the early appearance of Raynaud’s symptoms will be followed, sometimes years later, by their ANA becoming positive and additional scleroderma symptoms developing over time.
The first specific clinical symptom to suggest a diagnosis of scleroderma is skin thickening that begins as swelling or “puffiness” of the fingers and hands. The puffiness is usually worse in the morning and reduces later in the day, especially in early stages of the disease. Later the skin becomes hard, shiny, and leathery. With diffuse scleroderma, these areas of hardness are widespread and typically appear on both sides of the body. In the more limited form, skin thickening is often restricted to the hands, feet, and face. Eventually, tissue loss occurs and the skin becomes more highly colored.
People with limited scleroderma usually have Raynaud’s symptoms for years (often 5 to 10 years) before other signs of scleroderma are noted. However, even the limited form can, in rare cases, present with internal organ involvement without being preceded by Raynaud’s symptoms. Patients with limited scleroderma are less likely to develop severe lung, heart, or kidney involvement than patients with diffuse disease, although all of these complications can occur late in the disease process. Many patients with limited scleroderma eventually develop a cluster of symptoms that are listed using the acronym CREST – the old name for limited scleroderma. CREST is an acronym derived from the syndrome’s five most prominent symptoms:
- C – calcinosis, painful calcium deposits in the skin. It presents as small, localized, hard masses on fingers, forearms, or other pressure points.
- R – Raynaud’s phenomenon. Raynaud’s phenomenon is characterized by the intermittent loss of blood to various parts of the body – particularly the fingers, toes, nose, and/or ears after exposure to cold and causes tingling sensations, numbness, and/or pain. This can result in ulceration and necrosis of the fingertips and in some severe cases, lead to amputation of the affected digits.
- E – esophageal dysfunction, reflux (heartburn), difficulty swallowing caused by internal scarring. If the heartburn symptoms are not well controlled, the repeated acid exposure can eventually lead to a condition known as Barrett’s esophagus, a pre-cancerous condition. The esophagus may eventually have areas that are narrowed so that swallowing may become difficult. The small intestine may also lose the ability to push food through to the large intestine leading to malabsorption and increased bacterial growth in the small intestine.
- S – sclerodactyly, thickening and tightening of the skin on the fingers and toes.
- T – telangiectasia, red spots on the hands, palms, forearms, face and lips.
Depending on the particular antibody profile for patients diagnosed with limited scleroderma, they can be at increased risk of developing kidney failure, lung fibrosis, or pulmonary artery hypertension, but these complications usually occur at a much later date than with diffuse scleroderma. Severe fatigue and muscle pain are also very common symptoms.
With diffuse scleroderma, there is usually a short interval (weeks or months) between the development of Raynaud’s and significant additional symptoms, and, in some cases, Raynaud’s will not be the first symptom. Relatively rapid skin changes often occur in the first few months of the disease and continue to progress over the next 2 to 3 years. This is often followed by a partial remission of the skin changes, and the skin either thins or sometimes returns toward normal thickness. The severe fibrosis of the skin, especially in the fingers and hands, can cause significant disability. Diffuse scleroderma can also include a wide range of potential complications, including painful inflammation of the muscles, severe fatigue, swelling of the fingers and/or hands, gastrointestinal problems, lung fibrosis, pulmonary artery hypertension, progressive kidney failure, and cardiovascular problems. Internal organ involvement often occurs early in diffuse scleroderma and can be the initial presenting symptom.
Detailed information on scleroderma symptoms can be found in the Scleroderma FAQ.