This section includes a list of possible symptoms that can occur with different forms of systemic scleroderma. No patient will experience all of these symptoms and, even among patients who have the same specific subtype of systemic scleroderma, there is a tremendous variability in terms of which symptoms ultimately will occur and in what order.
Clinical Features – General
Scleroderma often begins with Raynaud’s phenomenon (see below) – the fingers and sometimes the toes lose circulation and turn white upon exposure to cold. Raynaud’s phenomenon usually (but not always) precedes skin changes by several months with diffuse scleroderma and often precedes skin changes by several years with limited scleroderma. Other early symptoms may be painful joints, morning stiffness, red swollen hands, fatigue, and/or weight loss. It is important to note, however, that Raynaud’s phenomenon without any underlying disease is not uncommon in the general population, especially among young women. This form of Raynaud’s is called “primary Raynaud’s.” A key distinguishing characteristic is that with primary Raynaud’s, the anti-nuclear antibody (ANA) will normally be negative, while with Raynaud’s which accompanies scleroderma or other auto-immune disorders (secondary Raynaud’s), ANA is usually positive. The clear majority of young women with Raynaud’s symptoms that appear in their teenage years never develop a positive ANA or any systemic damage or skin changes. However, in a small percentage of this population, the early appearance of Raynaud’s symptoms will be followed years later by ANA becoming positive and additional scleroderma symptoms developing over time.
The first specific clinical symptom to suggest a diagnosis of scleroderma is skin thickening that begins as swelling or “puffiness” of the fingers and hands. The puffiness is usually worse in the morning and reduces later in the day, especially in early stages of the disease. Later the skin becomes hard, shiny, and leathery. With diffuse scleroderma, these areas of hardness are widespread and typically appear on both sides of the body. In the more limited form, skin thickening is often restricted to the hands and face. Eventually, tissue loss occurs and the skin becomes more highly colored.
People with limited scleroderma usually have Raynaud’s symptoms for years (often 5 to 10 years) before other signs of scleroderma are noted. However, even the limited form can, in rare cases, present with internal organ involvement without being preceded by Raynaud’s symptoms. Patients with limited scleroderma are less likely to develop severe lung, heart, or kidney involvement than patients with diffuse disease, although these complications can occur late in the disease process. (The likelihood of developing specific complications based on antibody type is shown in Table 2.) Many patients with limited scleroderma eventually develop a cluster of symptoms that are listed using the acronym CREST. CREST is an acronym for calcinosis, Raynaud’s phenomenon, esophageal dysfunction, sclerodactyly and telangiectasia. Calcinosis is the abnormal accumulation of calcium salts under the skin and in many other organs. It presents as small, localized, hard masses on fingers, forearms, or other pressure points. Raynaud’s phenomenon is characterized by the intermittent loss of blood to various parts of the body – particularly the fingers, toes, nose, and/or ears after exposure to cold and causes tingling sensations, numbness, and/or pain. This can result in ulceration and necrosis of the fingertips and in some severe cases, lead to amputation of the affected digits. Dysfunction of the lower esophagus results in chronic heartburn and possible esophageal scarring. If the heartburn symptoms are not well controlled, the repeated acid exposure can eventually lead to a condition known as Barrett’s esophagus, a pre-cancerous condition. The esophagus may eventually have areas that are narrowed and swallowing may become difficult. The small intestine may also lose the ability to push food through to the large intestine leading to malabsorption and increased bacterial growth in the small intestine. Sclerodactyly, a condition in which the skin becomes thin, shiny, and bright, results in decreased function of the fingers and toes. Telangiectasia, the appearance of small blood vessels near the surface of the skin, usually on the face, hands, and in the mouth, is unsightly but not debilitating. Depending on the antibody profile for patients diagnosed with limited scleroderma, they can be at increased risk of developing kidney failure, lung fibrosis, and pulmonary artery hypertension, but these complications usually occur at a much later date than with diffuse scleroderma.
With diffuse scleroderma, there is usually a short interval (weeks or months) between the development of Raynaud’s and significant additional symptoms, and, in some cases, Raynaud’s will not be the first symptom. Relatively rapid skin changes often occur in the first few months of the disease and continue to progress over the next 2 to 3 years. This is often followed by a remission of the skin changes, and the skin either thins or sometimes returns toward normal thickness. The severe fibrosis of the skin, especially in the fingers and hands, can cause significant disability. Diffuse scleroderma can also include a wide range of potential complications, including inflammation of the muscles, swelling of the fingers and/or hands, microvascular abnormalities, gastrointestinal malfunction, lung fibrosis, pulmonary artery hypertension, progressive kidney failure, and cardiovascular problems. Internal organ involvement often occurs early in diffuse scleroderma and can be the initial presenting symptom.
Raynaud’s Phenomenon
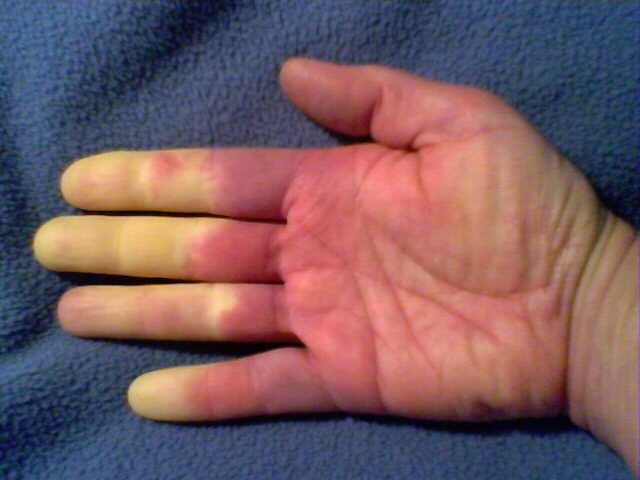


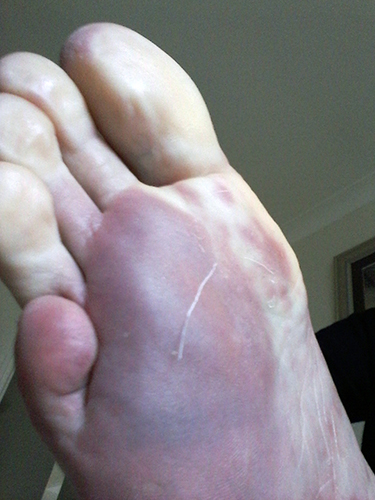
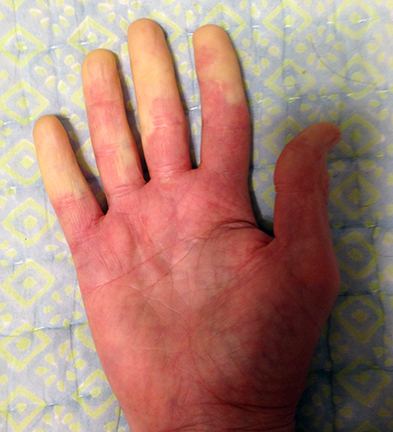
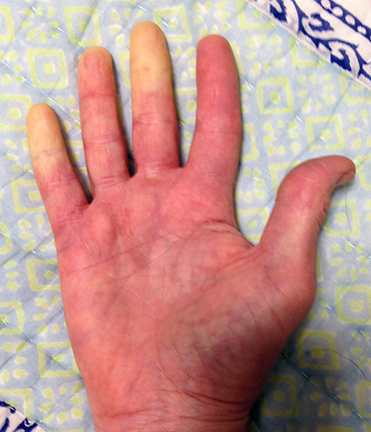
Raynaud’s phenomenon is characterized by cold sensations and color changes in the hands and feet. Upon exposure to cold or emotional stress, the fingers and/or toes (sometimes the nose), lose circulation and turn white (blanch). Once the digits are re-warmed the blood flow returns, commonly 10 to 15 minutes later. The affected portion of the digits will often turn a bluish color or will appear mottled before returning to normal appearance.
Several studies have reported that between 4% and 15% of the general adult population, primarily women, have symptoms of Raynaud’s phenomenon. These symptoms are usually quite mild and are not associated with any underlying disease. This form of Raynaud’s is known as “primary Raynaud’s.” It is also associated with a negative ANA. It generally first appears at a much younger age than secondary Raynaud’s, often before the age of 20. When Raynaud’s attacks are intense or long lasting or first occur after the age of 20, there is an increased likelihood that the Raynaud’s is secondary to an underlying autoimmune disorder. Note that in addition to scleroderma, Raynaud’s can be associated with a number of other disorders, for example, lupus, mixed connective tissue disorder, polymyositis, dermatomyositis, cold agglutinin disease, or hypothyroidism.
With secondary Raynaud’s there will usually be enlargement of the blood vessels at the base of the fingernails (nail bed capillary enlargement), although this is not always the case.
Skin Changes
In the earliest stages of scleroderma, the skin appears mildly inflamed with swelling and often redness. The skin gradually thickens (more rapidly in the diffuse form) and the patient feels progressive “tightening” of the skin with decreased flexibility. The skin changes are more widespread in the diffuse form and the skin can become “hyperpigmented,” giving the skin a salt and pepper appearance.
The pattern of skin changes is different for limited scleroderma and diffuse scleroderma. With limited scleroderma, the skin changes are typically limited to the fingers and lower arms, toes and lower legs, and the face. With diffuse scleroderma, the changes can cover more of the body including upper arms and legs and the trunk area.
As the skin changes progress, the skin becomes thicker and can become severely dried with intense itching. This stage can progress for a long period, up to several years. With diffuse scleroderma, the inflammation and further thickening stops as the skin begins to thin, although the skin will usually bind with underlying structures. Painful ulcerations can occur at joints.
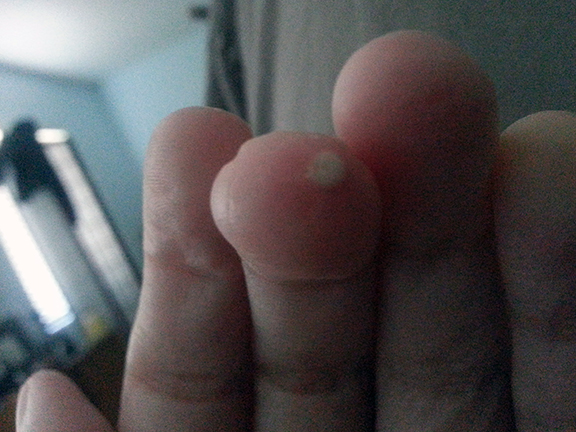
With the limited form of the disease calcium deposits may form under the skin. These can appear as white spots or ulcerations and may be quite painful. Spider veins (telangiectasia) often appear on the fingers, chest, face, lips, and tongue.
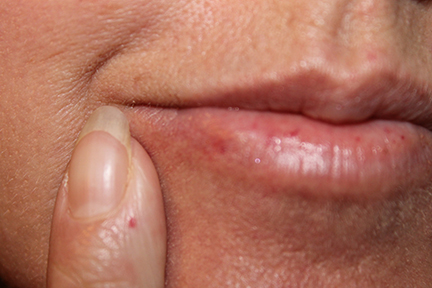
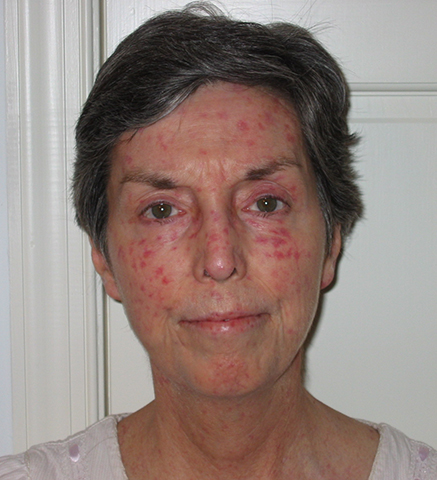
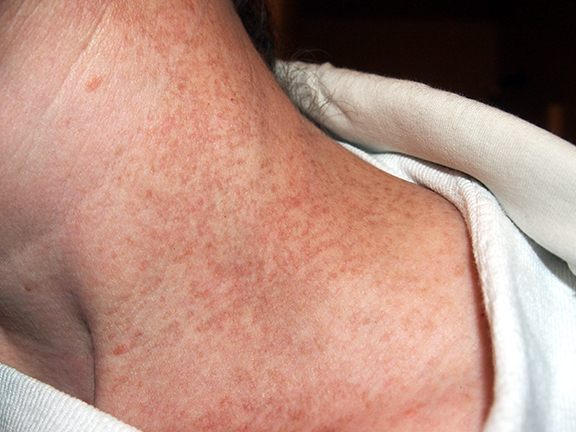
Telangiectasias
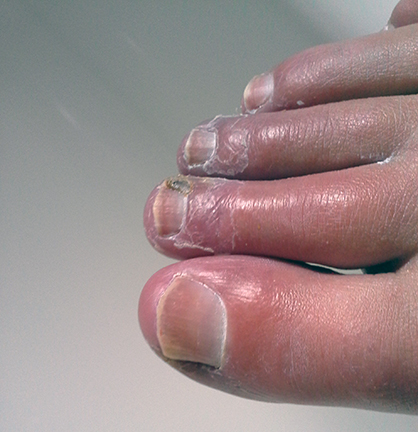
Calcinosis
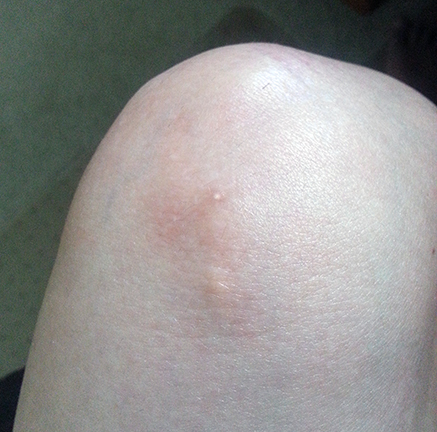
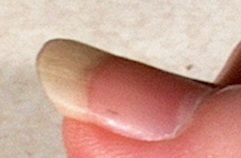
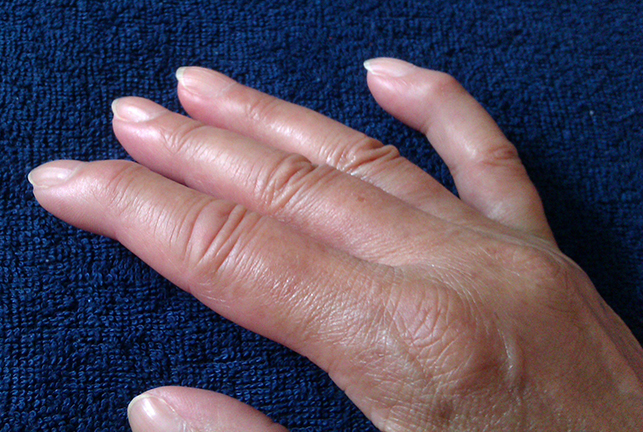
Puffy Fingers


Miscellaneous
Musculoskeletal (Muscles and Joints)

Nonspecific muscle pain and stiffness are often some of the earliest symptoms of scleroderma. While arthritis can also occur, the pain and stiffness over the joints is greater than would normally be expected based on the degree of inflammation visible. Pain can also occur along tendons and into muscles of the arms and legs. This can occur with movement of the ankles, wrists, knees, or elbows. These symptoms are more common in the diffuse form of the disease.
Often, a grating sound can be heard as the inflamed tissues move over each other, particularly at and below the knees. With diffuse scleroderma, the fingers, wrists, and elbows can become fixed in flexed positions because of the scarring of the skin. In the limited form, this is usually limited to the fingers.
In later stages of the disease, muscle loss and weakness are the main problems. In some cases, however, some of the symptoms may be caused by some of the drugs commonly used to treat scleroderma, such as steroids.
Recent research has isolated which subtypes of Scleroderma are more likely to have muscle problems. This is covered in Table 2 on our Scleroderma Antibodies and Clinical Relevance page.
Pulmonary (Lungs)

Some reduction of lung functioning occurs in almost all cases of scleroderma, both in the limited and diffuse forms. However, unless closely monitored, there may be no symptoms until later stages of the disease, at which point lung problems can become a major cause of death. The most common initial symptom is shortness of breath after exercise or other exertion. Later, a persistent non-productive cough can develop. Usually, there is no chest pain caused by the lung involvement, although chest pain can occur from other causes such as muscle pain or heartburn.
There are two different lung complications that are associated with scleroderma – interstitial lung disease (ILD) and pulmonary artery hypertension (PAH). While these can both occur, there is a definite association between different subtypes of scleroderma and the likelihood of developing these specific complications (see Table 2).
Interstitial lung disease (ILD) is basically inflammation and scarring of the lung tissue caused by progressive fibrosis of the lungs. Pulmonary artery hypertension means high blood pressure in the lungs. Patients with limited scleroderma have the greater risk of developing progressive blood vessel narrowing in the lungs even in the absence of lung scarring and inflammation.
The main diagnostic tool for lung problems is a pulmonary function test (PFT), often done on a yearly basis. Two different measures are specifically looked at when diagnosing lung problems: forced vital capacity (FVC) and diffusion capacity of the lung for carbon (DLCO). With ILD, these two measures tend to decline simultaneously while with PAH the DLCO measure declines more rapidly. High-resolution computerized axial tomography (CAT scan) is more useful for diagnosis of ILD than a standard x-ray. For detection of PAH, Doppler echocardiography is useful and should also be done on an annual basis for patients who are likely to develop this complication (based on specific antibody subtypes). An additional method of evaluating the severity of PAH, as well as the effectiveness of PAH treatments is a simple test called the 6-minute walk test (6MWT). Basically, this test measures the distance that a patient can walk on a flat, hard surface in a period of 6 minutes. This test is inexpensive and easy to perform and assesses distance walked, shortness of breath, and O2 saturation levels. Several studies have shown that the score on the 6MWT is correlated with mortality rates over a one to three-year period.
While the course of lung involvement is highly variable, most patients have an early but limited decline in lung functioning and then either stabilize or improve. About one third of patients have a more severe progression for several years before stabilizing. While other lung problems can develop secondary to other complications, these are much less common. In addition, there is an increased risk of lung cancer with scleroderma.
Gastrointestinal

Some of the most common symptoms of scleroderma are various difficulties with the gastrointestinal tract. This occurs with both systemic forms of the disease. As fibrosis develops in the upper part of the gastrointestinal tract, moderate to severe heartburn commonly develops. At later stages, the muscles that propel food from the mouth to the stomach function less efficiently leading to difficulty in swallowing. In addition, the stomach may empty more slowly adding to heartburn symptoms and causing bloating, nausea, and vomiting. Some scleroderma patients develop what is called “watermelon stomach” (GAVE syndrome), in which the stomach develops red streaked areas from widened blood vessels. This can increase the risk of stomach cancer and can lead to anemia (low red blood cell counts).
There is some research that supports the idea that patients with severe reflux disease may breathe in tiny amounts of stomach acid that in turn may contribute to lung scarring and fibrosis. Untreated or undertreated reflux can lead to erosion of the esophagus resulting in bleeding, strictures with narrowing of the esophageal opening, and Barrett’s esophagus, a pre-cancerous condition.
In the lower part of the GI tract, movement of the food through the intestines can be slowed, sometimes resulting in an increase of bacterial levels in the intestines and reduction in food absorption. This can cause weight loss, cramping, constipation or diarrhea, and in severe cases, malnutrition. An under-reported symptom that can develop with severe scleroderma is fecal incontinence (leakage) because of fibrosis and reduced muscle tone of the internal anal sphincter.
Two tests are often used to investigate the extent and severity of upper GI symptoms related to systemic scleroderma: upper endoscopy and esophageal manometry.
- Upper Endoscopy involves using a thin scope with a tiny light and camera that is inserted through the mouth to examine the condition of the esophagus, stomach, and the first part of the small intestine. This is done under sedation in a hospital setting and allows the physician to detect a number of potential Scleroderma related complications, including esophageal erosion from frequent reflux, Barrett’s esophagus, and watermelon stomach.
- Esophageal Manometry is a procedure for measuring how well the muscles of the esophagus work, especially the strength of the lower esophageal sphincter. The procedure is performed by passing a thin plastic tube through one nostril, down the throat, and into the esophagus. Once the tube is inserted, pressure readings can be done when the esophagus is resting or when the patient is swallowing. The strength of contractions while the patient is swallowing can help to diagnosis a number of swallowing related problems, and the measure of LES pressure can be useful in determining appropriate treatments for the patient’s reflux problems.
Cardiac (Heart)

Most systemic scleroderma patients have limited heart problems that may be detectable but are not clinically significant. Even with diffuse scleroderma, serious heart complications are uncommon and occur in only 10% to 15% of patients, usually within the first few years after the disease begins. The complication rate with limited scleroderma is even lower. When more severe heart problems develop, they can be difficult to manage and may be associated with poor prognosis. The most direct effect on the heart is scarring, which increases the risk of heart rhythm problems. Also, a condition called pericarditis (inflammation of the membrane around the heart) can occur.
Renal (Kidney)
Kidney involvement is common in scleroderma, although there may be no obvious clinical problems. Kidney problems tend to be more serious and more common in the diffuse form of the disease, especially with RNA Polymerase III antibodies, with life-threatening scleroderma renal crisis occurring in 10% to 20% of diffuse scleroderma patients. Scleroderma renal crisis is much less common in limited scleroderma although it can occur, often early in the disease. Approximately 80% of all major kidney problems occur within the first 4 to 5 years of the disease. For unknown reasons, serious kidney problems are more common in men and with patients who had an older age of disease onset. Note that treatment with high dose corticosteroids can increase the chances of developing major kidney problems and should generally be avoided in patients with early diffuse scleroderma.
Since systemic scleroderma patients tend to have relatively low blood pressure compared to the general population, any sudden increase in blood pressure is of concern with scleroderma patients. For this reason, frequent monitoring of blood pressure is important, especially for diffuse scleroderma patients for the first few years of the disease.
Sexual Dysfunction

Sexual dysfunction is very common in patients with systemic scleroderma. A recent study (Schouffoer et al. 2009) found that women with systemic sclerosis reported significantly impaired sexual functioning and more sexual distress than healthy controls, often leading to marital distress and depressive symptoms. Major problems were increased vaginal dryness, skin tightness and decreased lubrication resulting in painful intercourse, heartburn and reflux during intercourse, and reduced frequency and intensity of orgasms.
Men with systemic scleroderma are much more likely to have problems with erectile dysfunction (ED) than men with rheumatoid arthritis (Hong et al. 2004). Onset of ED averaged about three years after disease onset. In the Hong study, about 81% of men reported problems with ED compared to 48% with rheumatoid arthritis.
Other Symptoms

While scleroderma does not appear to cause major central nervous system dysfunction, recent studies have shown that more than 50% of all scleroderma patients develop moderate to major depression (Thombs et al. 2007). Patients also frequently have difficulty with altered self-image because scleroderma can be disfiguring in some cases. The incidence of depression is somewhat higher than would be expected in a population of patients with a severe, chronic disease. However, in almost all cases the depression is responsive to treatment with medications commonly used to treat depression.
It is very common for patients with both limited and diffuse forms of scleroderma to have severe, sometimes debilitating fatigue. It is not clear what the specific mechanism of action is for this fatigue, but anemia can often develop with scleroderma, which may contribute to the severity of this symptom.
A significant number of scleroderma patients also suffer from Sjögren’s syndrome (also called Sicca syndrome). The primary symptoms are dry mouth and eyes. This can result in dental complications and the need to use lubricating eye drops to prevent eye problems.
Hypothyroidism (reduced function of the thyroid) is very common in systemic scleroderma because of either fibrosis of the thyroid or thyroid autoimmune disorder. Hypothyroidism causes many bodily functions to slow down. Some of the more common symptoms include: hoarse voice, slowed speech, eye and face puffiness, weight gain, cold intolerance, dry skin, carpal tunnel syndrome, and coarse, dry, sparse hair.
Sleep disturbance is also common with scleroderma patients (Frech et al. 2011). There is a variety of reasons for this, including muscle and other pain (e.g., digital ulceration), difficulty breathing, and reflux symptoms.
Other symptoms that have been linked to scleroderma include: severe chronic chilling even in the absence of hypothyroidism, trigeminal neuralgia (sudden painful spasms in the lower portion of the face radiating to the neck), osteoporosis, increased occurrence of vertigo (dizziness), and liver damage. However, in some cases, the linkage may not necessarily be a direct result from the underlying disease process in scleroderma. For example, while an increased risk of osteoporosis is often listed as a potential complication of scleroderma, other co-factors such as early menopause, usage of corticosteroids, or malabsorption may in fact be the causal agent, rather than the disease process itself.